บททบทวนวารสาร
Review Article
Interstitial Lung Diseases in the Idiopathic Inflammatory Myopathies
Intira Masayavanich, M.D.
Fellow-in-Training
Division of Respiratory Disease and Tuberculosis
Department of Medicine
Faculty of Medicine Siriraj Hospital
Mahidol University
Introduction
Idiopathic inflammatory myopathies or myositis (IIM) are heterogeneous disorders characterized by varying degrees of muscle weakness and inflammation[1]. Lungs are the most common extramuscular involvement in IIM including respiratory muscle weakness, pulmonary hypertension, interstitial lung disease, and pleural effusion[2-3]. Interstitial lung disease (ILD) is the hallmark of pulmonary involvement that causes significant morbidity and mortality[2].
Classification of Idiopathic inflammatory myopathies
The major subgroups in adult IIM are dermatomyositis (DM), polymyositis (PM), and inclusion body myositis (IBM). Several diagnostic criteria for IIM have been proposed as shown in Table 1[4-7]. The Bohan and Peter’s criteria is the most widely used, but have several limitations including no external validation with the estimation of sensitivity or specificity and lack of specific criteria for exclusion of other forms of myopathies[8]. Recently, The European League Against Rheumatism/American College of Rheumatology (EULAR/ACR) published the classification criteria using multidisciplinary consensus that showed higher sensitivity and specificity for diagnosis of major subgroups of IIM[9].
Types of pulmonary involvement
Pulmonary involvement in IIM has various symptoms and signs that can be classified as direct pulmonary involvement (e.g. ILD and pulmonary hypertension) and indirect pulmonary complications (e.g. infection and respiratory muscle weakness) that are listed in Table 2[1,10]. Pulmonary involvement is common in PM and DM, but IBM has no direct pulmonary involvement. ILD is the most common pulmonary involvement that can cause significant morbidity and mortality. This article will focus on ILD-associated with PM and DM.
Table 1. Diagnostic criteria for idiopathic inflammatory myopathies
| Bohan and Peter’s criteria[4-5] | ENMC criteria[6] | Dalakas and Hohlfeld[7] |
| Features | Features (except inclusion body myositis) | Features |
| Symmetrical proximal muscle weaknessElevated serum muscle enzymesEMG consistent with myopathyMuscle biopsy with characteristic featuresTypical rash | Clinical criteria: subacute or insidious onset, pattern of weakness: symmetric proximal > distal, neck flexor > neck extensor, or rash typical of DM Elevated serum creatinine kinase level Other laboratory criteria (EMG, MRI, or MSAs detected in serum) Muscle biopsy (inclusion and exclusion criteria) | Subacute proximal muscle weakness Elevated serum muscle enzymes Muscle biopsy Typical rash |
| Diagnosis | Diagnosis | Diagnosis |
| Definite PM: 1-4 criteria present Probable PM: Any 3 of 1-4 present Possible PM: Any 2 of 1-4 present Definite DM: Rash + any 3 of 1-4 | Definite PM: 1 without rash + 2 + 4 Probable PM: 1 without rash + 2 + 3 (1 of 3) + 4 Definite DM: 1 + 4 Probable DM: 1 + 4 or 3 (1 of 3) Amyopathic dermatomyositis, possible dermatomyositis sine dermatitis, non-specific myositis, immune-mediated necrotizing myopathy diagnostic criteria: not shown | Definite PM: criteria 1, 2 with muscle biopsy showing inflammation with CD8/MHC-I complex and no vacuoles Probable PM: criteria 1 and 2 with muscle biopsy showing MHC-I expression without T cells or vacuoles Definite DM: rash + muscle biopsy Probable DM: no rash + typical biopsy ADM: Rash without muscle weakness |
Table 2. Pulmonary involvement in idiopathic inflammatory myopathies
| Pulmonary involvement | Diseases or conditions |
| Direct pulmonary involvement | Interstitial lung diseasePulmonary arterial hypertensionPulmonary hypertension due to chronic hypoxemia |
| Indirect pulmonary complication | Pulmonary infectionRespiratory muscle weaknessDrug-induced lung diseaseLung cancer |
Epidemiology
The prevalence of ILD-associated with PM/DM is ranging from 17 to 36%11-14. This variation is due to a lack of standardized criteria and screening for ILD and the limitation of retrospective studies10. The most common type of ILD in IIM patients is non-specific interstitial pneumonia (NSIP) follow by organizing pneumonia (OP) and usual interstitial pneumonia (UIP) as shown in Table 3.
Table 3. Interstitial lung disease in idiopathic inflammatory myopathies.
| Type | Frequency |
| Nonspecific interstitial pneumonia (NSIP) | +++ |
| Organizing pneumonia (OP) | ++ |
| Usual interstitial pneumonia (UIP) | ++ |
| Acute interstitial pneumonia (diffuse alveolar damage) | + |
| Lymphocytic interstitial pneumonia (LIP) | +/- |
Pathogenesis
The pathogenesis of ILD-associated with PM/DM remains unknown. It is generally accepted that genetically susceptible individuals and the exposure to some environmental factors like viral infection, smoking, inhalation of organic and inorganic dust may play a role. There were associated with human leukocyte antigen (HLA) class II, HLA-DRB*03, HLA- DQA1*05, and HLA-DQB1*02 irrespective of myositis subtype or anti-Jo-1 autoantibodies10. Immuno-histochemistry on muscle biopsy provides some data about immune-mediated mechanisms[1].
In polymyositis, CD8+ T-cells are usually found with diffuse cytotoxic effect leading to muscle cell necrosis predominantly within the endomysium of healthy-appearing, non-necrotic muscle fibers expressing major histocompatibility complex (MHC) class I antigen. In DM, B-cells and CD4+ T-cells were found in perivascular areas and complement on the endothelial cell wall of endomysial vessels which may be responsible for complement-mediated microangiopathy in patients with DM[15].
Clinical presentation
Clinical presentation is variable including asymptomatic (25%), subacute or chronic ILD (58%), and rapidly progressive ILD (17%)[2,13]. Progressive dyspnea, non-productive cough and decreased exercise tolerance are the most common symptoms that can precede (19%), occur concomitantly with (42%), or present after (39%) the diagnosis of PM/DM [2,10,13]. However, these symptoms are non-specific to ILD because they can either present in patients with pulmonary involvement other than ILD, e.g. respiratory muscle weakness and pulmonary infection[16]. Physical examination usually reveals velcro crackles with or without a sign of respiratory distress or central cyanosis depending on the severity of disease. Older age (>45 years old), joint symptoms, and positive anti-Jo-1 at onset predict the presence of ILD in IIM patients[17].
Asymptomatic or occult ILD
Patients do not have respiratory symptoms. ILD is identified on radiological imaging, including plain radiographs or high-resolution computed tomography of the chest (HRCT) or abnormal pulmonary function tests18.
Subacute or chronic ILD
Chronic form of ILD was defined as a slowly progressive presentation with gradual deterioration over more than 3 months19. Patients with slow progressive pattern had a better survival rate than patients presented with rapidly progressive ILD regardless of the underlying IIM[19].
Rapidly progressive ILD
Rapidly progressive ILD is an acute interstitial pneumonia (AIP) that develops in several weeks or a few months[10]. The presentation includes fever, dyspnea and rapid progression to acute respiratory failure with abnormal chest X-ray. The acute and aggressive presentation is more frequently seen in dermatomyositis, clinically amyopathic dermatomyositis (CADM), and hypomyopathic dermatomyositis[2,12, 20-21]. This form of aggressive ILD is more resistant to treatment with intensive immunosuppressive therapy and has a very poor prognosis with high mortality[2,10,13]. A meta-analysis demonstrated that the presence of anti-MDA5 autoantibodies in DM patients increased the risk of developing rapidly progressive ILD with a sensitivity of 77% and a specificity of 86% [16, 22]. The detection of anti-aminoacyl-tRNA-synthetase (anti-ARS) autoantibodies seems to be a protective factor against this presentation[23].
Antisynthetase syndrome
The antisynthetase syndrome is characterized by the presence of one of antisynthetase antibodies in combination with fever (43%), polyarthritis (62%), myositis (57%), ILD (70%), mechanic’s hands (28%), and Raynaud’s phenomenon (47%) as shown in Table 41. This syndrome occurs in up to one-third of patients with PM and DM. ILD is found in up to 70% of patients with antisynthetase syndrome and may precede the other symptoms[10]. Joint manifestations range from polyarthralgia to destructive polyarthritis of the hands, wrists, elbows, and knees. Erosive joint disease is uncommon[1]. 5–8% of antisynthetase syndrome manifests as overlap diseases with other connective tissue diseases, e.g. rheumatoid arthritis, systemic lupus erythematosus, scleroderma, and Sjögren’s syndrome[24]. ILD dominates the prognosis of antisynthetase syndrome which is associated with more than 40% mortality. Anti-Jo-1 and anti-PL are autoantibodies to histidyl-tRNA-synthetase which is the most common autoantibodies found in 60-80% of the antisynthetase syndrome. The other autoantibodies are found in 20-40%: anti-PL7 (10-15%), anti- PL12 (5-10%), anti-EJ, anti-OJ, and anti-KS (5-15%)10. Anti-PL7 and anti-PL12 seem to be associated with isolated pulmonary fibrosis or arthritis[25].
Table 4. Proposed criteria for myositis associated anti-tRNA synthetase antibody[1]
| Features |
| Positive serological tests for anti-tRNA synthetase antibody plus one major involvement:Evidence of overt or hypomyopathic myositis by Bohan and Peter critieria*Evidence of interstitial lung disease accoding to ATS critieriaEvidence of articular involvement** |
| Or two minor involvement:Unexplained persistent feverRaynaud’s phenomenonMechanic’s hands |
* Elevated creatine phosphokinase (CPK) levels, myalgia, proximal muscle weakness, positive muscular biopsy, electromyographic triad of myositis or MRI muscular edema
** Symmetrical inflammatory arthralgia or overt arthritis
Anti-melanoma differentiation-associated gene 5 (MDA5)-related ILD
In 2005, anti-MDA5 autoantibody, originally called anti-CADM-140, was firstly described in 8 patients with CADM of whom 50% had developed rapidly progressive ILD [25-27]. The anti-MDA5 autoantibodies are specific to DM which may be found in 7-13% of DM patients. Significant myositis could be found in only 20% of patients. Skin manifestations are digital ulcers which usually located over the Gottron’s papules, digital pulps, periungual area, or other sites of dermatomyositis rash. Other manifestations include panniculitis, arthritis (80%), Raynaud’s phenomenon (45%), and mechanic’s hands (80%). The screening for ANA may be negative. Other laboratory investigations are a high level of serum ferritin, alpha-glutamyl transpeptidase, and lower CK values at presentation[27].
Several studies have reported that anti-MDA5 had a strong association with rapidly progressive ILD and resulted in poor survival[25,28]. The survival rate of patients as low as 54% at 6 months. An autoantibodies titer level >500 U/mL is associated with treatment resistance and a higher risk of short-term death from acute respiratory failure. High serum ferritin (> 1500 ng/ml) has also been associated with poor survival[27].
Investigation
Chest imaging
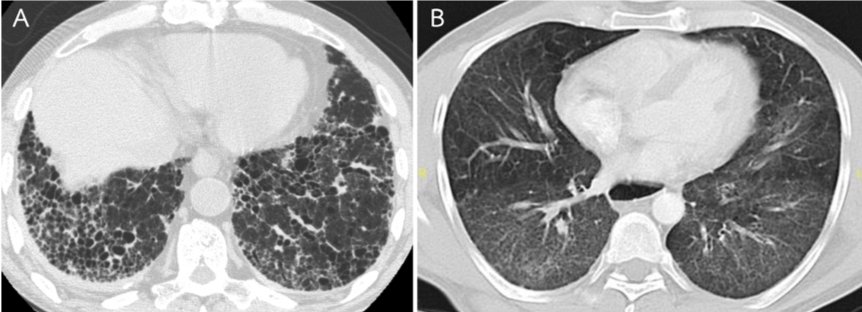
Chest X-ray is helpful but has limited sensitivity and specificity in the diagnosis of ILD. HRCT is more sensitive in the detection and characterization of ILD. The most common HRCT findings include consolidation, reticulation, ground-glass opacities, and peri-bronchovascular thickening as shown in Figure 1B and Table 5 [1]. Honeycombing, traction bronchiectasis, and bronchiolectasis are less frequently found (Figure 1A) [1,11]. A combination of reticulation and area of consolidation is usually found in acute to subacute onset of ILD.
HRCT patterns at diagnosis can predict the prognosis of patients. Dominant consolidation is generally responded well to corticosteroids and immunosuppressive agents. Dominant ground-glass opacities and/or reticulation without honeycombing, subpleural bands and traction bronchiectasis are often associated with severe ILD and a possibility of deadly outcome[1].
Table 5. Common HRCT and histopathologic patterns
| Pattern | HRCT findings | Histopathology |
| Organizing pneumonia (OP) | Consolidation (unilateral or bilateral), usually peripheral, subpleural, or peribronchovascular | Foci of granulation tissue in alveoli and their ducts |
| Non-specific interstitial pneumonia (NSIP) | Subpleural and bibasilar ground glass opacities with some traction bronchiectasis and reticulation | Temporally and geographically homogenous infiltration of lung interstitium by inflammatory cells with moderate collagen deposition, preserved lung architecture, and scarcity of fibroblastic foci and honeycombing |
| Usual interstitial pneumonia (UIP) | Subpleural and bibasilar honeycombing with reticulation and traction bronchiectasis | Spatially and temporally heterogenous collagen deposition within lung, architectural destruction, fibroblastic foci, honeycombing, and moderate inflammatory cell infiltration |
| Acute interstitial pneumonia | Diffuse ground glass opacities with consolidation with or without reticulation and traction bronchiectasis on background | Diffuse interstitial inflammation with edema and hyaline membrane production |
Pulmonary function tests (PFTs)
PFTs are a sensitive but non-specific test in the diagnosis of ILD but they can be used for evaluation of disease severity, course of disease and treatment response. Patients typically demonstrated a restrictive ventilatory defect and decreased diffusion capacity for carbon monoxide (DLCO). PFTs should be carefully interpreted due to a potential coexistence of respiratory muscle weakness and ILD which PFTs also show restrictive ventilatory defect from chest wall restriction and diffusion defect from basal lungs atelectasis[11]. FVC <60% predicted at the time of diagnosis is associated with poor survival.
Bronchoscopy
Bronchoscopy with bronchoalveolar lavage (BAL) is useful for the exclusion of occult infection and other diseases resembling ILD. Transbronchial lung biopsy (TBLB) may be helpful in the diagnosis of organizing pneumonia and ruling out other causes e.g. infection and malignancy but it has a limited role in the diagnosis of NSIP or UIP due to inadequate lung tissue.
Surgical lung biopsy
SLB in the diagnosis of ILD remained controversial.
Histopathology
Obtaining lung tissue for the diagnosis of ILD is challenging. According to the limitation of TBLB, transbronchial cryobiopsy (TBCB) and surgical lung biopsy (SLB) may play an important role in obtaining larger tissue for histopathological diagnosis. However, TBLB and SLB are more invasive procedures with reported major complications, e.g. pneumothorax, bleeding, acute exacerbation, and death. Currently, we recommend that TBLB and SLB should be performed only in patients with clinical and HRCT uncertainty.
The most common histology is NSIP and OP. Other histologic subtypes include usual interstitial pneumonia, diffuse alveolar damage, and lymphocytic interstitial pneumonia[2,11,18]. Several histopathologic patterns correlate with HRCT findings, therefore, a tissue biopsy may not be necessary if there are typical findings on HRCT as shown in Table 5[2].
Autoantibodies
The screening for ANA may be negative. Further testing for myositis-specific autoantibodies (MSA) and myositis-associated autoantibodies (MAA) is necessary. ILD is commonly found in patients with antisynthetase syndrome. DM patients with positive antisynthetase antibodies are more likely to have ILD than antibody-negative DM (94% and 23%, respectively) and more likely to require higher and prolonged doses of immunosuppressive agents. The most common antisynthetase antibody is anti-Jo-1 is the most common antisynthetase antibody which strongly associated with the presence of ILD (89%)[11].
Serum biomarkers
Several serum biomarkers have been studied, e.g. glycoprotein Krebs von den Lundgen-6 (KL-6), cytokeratin 19 fragment, and surfactant protein A and D, which showed variable results. Currently, there is limited data about the role of serum biomarkers in the diagnosis of ILD associated IIM and disease progression[2,29-30].
Treatment
1. Pharmacologic treatment
Corticosteroids is the mainstay treatment of IIM. However, there are no established treatment regimens for ILD associated with IIM [10-11]. Immunosuppressive agents should be considered when ILD is severe or rapidly progressive in order to reduce the side effects of corticosteroids and improve the response to treatment[18].
Corticosteroids
Corticosteroids are the first-line treatment of IIM with a starting dose of 0.75 to 1 mg/kg/d equivalent to prednisolone with a slowly tapering based on clinical response. Approximately half of patients have a good clinical response to initial corticosteroid therapy. Corticosteroids monotherapy achieved favorable responses in 37.5% of PM-ILD but only 8.3% in ILD with DM patients[26]. The overall 2.5-year survival rate of DM-ILD was 58% and the 5-year survival of PM-ILD patients was 81%. A combination with immunosuppressive drugs is recommended for steroid-sparing or increased efficacy, particularly in rapidly progressive ILD[10].
Azathioprine
Azathioprine is commonly used in ILD associated with IIM as a corticosteroid-sparing agent or maintenance therapy, solely or following induction with cyclophosphamide. Dosage ranges from 2-3 mg/kg/day[2,11].
Cyclophosphamide
Cyclophosphamide can be administered orally or monthly intravenous pulse in combination with corticosteroids. According to side effect profiles, cyclophosphamide is commonly used in rapidly progressive ILD and refractory ILD. Several case studies have demonstrated its potential efficacy in treating ILD associated with IIM. Dosage ranges from 1-2 mg/kg/day per oral or monthly intravenous pulse 300-800 mg/m2 at least 6 times in combination with prednisolone 0.5-1 mg/kg/day[2,18].
Methotrexate
Methotrexate is used as an adjunctive treatment in arthritis and myositis in PM/DM. However, there is controversy about the efficacy of methotrexate in the treatment of ILD associated IIM10. Methotrexate should be used with caution due to a known association with idiosyncratic drug-related hypersensitivity pneumonitis11. It is difficult to distinguish worsening symptoms after drug initiation between the manifestation of ILD and drug-induced lung disease11. It is usually administered orally at a dose of 0.2-0.3 mg/kg weekly.
Calcineurin inhibitors
Cyclosporin A and tacrolimus are the T-cell- and IL-2 production inhibitor29. Several case series have demonstrated that maintenance with cyclosporin A and tacrolimus may be an appropriate choice for early, slowly progressive, and non-diffuse ILD because of their safety profile and efficacy on stabilized lung functions[2,29]. Cyclosporin A dosage is 2-5 mg/kg/day adjusted for trough level of 100-200 ng/ml. Tacrolimus dosage depends on the trough level (5-20 ng/mL)[2].
Mycophenolate mofetil (MMF)
MMF has a potential efficacy in reversing progression or stabilization of disease activity in various connective tissue diseases associated with ILD including IIM. Several studies have demonstrated remission or stabilization of ILD in 80% of patients with chronic ILD. MMF in combination with corticosteroids is increasingly used due to its potential efficacy and safety profile. Recommended dose is 2,000-3,000 mg/day[2].
Plasmapheresis
Plasma exchange is used to remove circulating autoantibodies, cytokines, and immune complexes. Case reports of efficacy of plasmapheresis in patients with antisynthetase syndrome who were refractory to corticosteroids and other immunosuppressive therapies[18].
Intravenous immunoglobulin (IVIG)
IVIG is widely used for the treatment of numerous autoimmune diseases. The efficacy of IVIG in ILD associated with IIM is uncertain. There were some case series demonstrated an improvement of PFT and CT imaging following the treatment of IVIG in patients with refractory myositis[2,18].
Rituximab
Rituximab is a monoclonal antibody targeting the CD20 protein. The effectiveness on stabilizing and/or improving the pulmonary function tests, including FVC, DLCO, and TLC, is 72%. The most benefit on pulmonary function was observed in patients with disease duration <1 year and acute onset of ILD. Patients with antisynthetase syndrome, mainly positive anti-Jo-1 and anti-Mi-2, were more likely to respond to rituximab therapy[31]. There is an upcoming trial which is the first randomized control trial to study the efficacy of rituximab versus cyclophosphamide for treatment of connective tissue disease-associated interstitial lung disease as first-line treatment in CTD-associated ILD32. The recommended dose is 1,000 mg intravenously on day 0 and day [14].
2. Non-pharmacologic treatment and vaccination
Pulmonary rehabilitation
Currently, there is no recommendation about the criteria for pulmonary rehabilitation in ILD patients. Recent data demonstrated that exercise can improve muscle strength and reduce impairment in patients with IIM33. A prospective cohort study, which included patients with different types of ILD, described improvement in quality of life and 6-minute walk distance following pulmonary rehabilitation[34]. Although no study has evaluated the efficacy of pulmonary rehabilitation specifically in patients with myositis related ILD, it is likely that they would benefit from therapy[35].
Long-term oxygen therapy
No clinical trial for the use of long-term oxygen therapy in patients with ILD associated with IIM specifically. However, several guidelines recommend the use of oxygen in ILD patients who have significant hypoxemia, defined by oxygen saturation of <88% or partial pressure of O2 (PaO2) of <55 mmHg. Patients should be reassessed regularly and oxygen prescription should be adjusted as oxygen demand change.
Vaccination
Pulmonary infection can contribute to a worsening of symptoms and cause significant morbidity and mortality. There is no study evaluating the impact of vaccination on ILD patients. However, several guidelines recommend that ILD patients should receive influenza and pneumococcal vaccination.
Lung transplantation
There are very few published case reports or series of successful lung transplantation in patients with ILD related IIM36-38. There is a slightly lower 1-year and 2-year survival rates in patients with ILD related IIM (67.5% and 56.3%, respectively) compared to those of IPF (72.7% and 66.3%, respectively) and those of CTD-associated ILD (77.7% and 68.2%, respectively)[38].
Palliative and end-of-life care
The objective of palliative care is to provide comfort to patients and caregivers and to reduce the burden of symptoms. Discussion about diagnosis and prognosis of disease between physician, patient, and caregivers should be undertaken once the diagnosis is made.
Prognosis
The predictors of poor outcome include acute presentation, neutrophilic alveolitis, initial DLCO < 45%, FVC < 60%, DM, microangiopathy, digital infarcts in DM/ADM, and histopathologic diagnosis of UIP[2,19]. The survival of ILD associated with IIM was 94%, 90%, and 87% at 1, 3, and 5 years, respectively[2].
Conclusion
ILD is the most common pulmonary manifestation of IIM which causes significant morbidity and mortality. ILD associated IIM has various manifestations and clinical courses. Careful evaluation and investigation is necessary for diagnosis and treatment decision.
References
1. Lega J-C, Reynaud Q, Belot A, Fabien N, Durieu I, Cottin V. Idiopathic inflammatory myopathies and the lung. Eur Respir Rev 2015; 24:216-38.
2. Kalluri M, Oddis CV. Pulmonary manifestations of the idiopathic inflammatory myopathies. Clin Chest Med 2010; 31:501-12.
3. Broaddus VC MM, Ernst JD, et al. Murray and Nadel’s Textbook of Respiratory Medicine. 6th ed. Philadelphia: Elsevier Saunders; 2016.
4. Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). New Engl J Med 1975; 292:344-7.
5. Bohan A, Peter JB. Polymyositis and Dermatomyositis. New Engl J Med 1975; 292:403-7.
6. Hoogendijk JE, Amato AA, Lecky BR, et al. 119th ENMC international workshop: trial design in adult idiopathic inflammatory myopathies, except for inclusion body myositis, 10-12 October 2003, Naarden, The Netherlands. Neuromuscul Disord 2004; 14:337-45.
7. Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003; 362:971-82.
8. Lazarou IN, Guerne PA. Classification, diagnosis, and management of idiopathic inflammatory myopathies. J Rheumatol 2013; 40:550-64.
9. Lundberg IE, Tjarnlund A, Bottai M, et al. 2017 European League Against Rheumatism/American College of Rheumatology Classification Criteria for Adult and Juvenile Idiopathic Inflammatory Myopathies and Their Major Subgroups. Arthritis Rheum 2017; 69:2271-82.
10. Labirua A, Lundberg IE. Interstitial lung disease and idiopathic inflammatory myopathies: progress and pitfalls. Curr Opin rheumatol 2010; 226:633-8.
11. Connors GR, Christopher-Stine L, Oddis CV, Danoff SK. Interstitial lung disease associated with the idiopathic inflammatory myopathies: what progress has been made in the past 35 years? Chest 2010; 138:1464-74.
12. Fathi M, Dastmalchi M, Rasmussen E, Lundberg IE, Tornling G. Interstitial lung disease, a common manifestation of newly diagnosed polymyositis and dermatomyositis. Ann Rheum Dis 2004; 63:297-301.
13. Marie I, Hachulla E, Cherin P, et al. Interstitial lung disease in polymyositis and dermatomyositis. Arthritis Rheum 2002; 47:614-22.
14. Selva-O’Callaghan A, Labrador-Horrillo M, Munoz-Gall X, et al. Polymyositis/dermatomyositis-associated lung disease: analysis of a series of 81 patients. Lupus 2005; 14:534-42.
15. Dalakas MC. Inflammatory Muscle Diseases. New Engl J Med 2015; 372:1734-47.
16. Chen Z, Cao M, Plana MN, Liang J, et al. Utility of anti-melanoma differentiation-associated gene 5 antibody measurement in identifying patients with dermatomyositis and a high risk for developing rapidly progressive interstitial lung disease: a review of the literature and a meta-analysis. Arthritis Care Res 2013; 65:1316-24.
17. Chen IJ, Jan Wu YJ, Lin CW, et al. Interstitial lung disease in polymyositis and dermatomyositis. Clin Rheuma 2009; 28:639-46.
18. Kawasumi H, Gono T, Kawaguchi Y, Yamanaka H. Recent Treatment of Interstitial Lung Disease with Idiopathic Inflammatory Myopathies. Clin Med Insights Circ Respir Pulm Med 2015; 9(Suppl 1):9-17.
19. Fujisawa T, Hozumi H, Kono M, et al. Prognostic factors for myositis-associated interstitial lung disease. PloS One 2014; 9:e98824.
20. Ye S, Chen XX, Lu XY, et al. Adult clinically amyopathic dermatomyositis with rapid progressive interstitial lung disease: a retrospective cohort study. Clin Rheumatol 2007; 26:1647-54.
21. Ito M, Kaise S, Suzuki S, et al. Clinico-laboratory characteristics of patients with dermatomyositis accompanied by rapidly progressive interstitial lung disease. Clin Rheumatol 1999; 18:462-7.
22. Gerfaud-Valentin M, Ahmad K, Piegay F, et al. [Interstitial lung disease-associated with amyopathic dermatomyositis and anti-MDA5 autoantibodies]. Rev Mal Respir 2014; 31:849-53.
23. Tillie-Leblond I, Wislez M, Valeyre D, et al. Interstitial lung disease and anti-Jo-1 antibodies: difference between acute and gradual onset. Thorax 2008; 63:53-9.
24. Imbert-Masseau A, Hamidou M, Agard C, Grolleau JY, Cherin P. Antisynthetase syndrome. Joint Bone Spine 2003; 70:161-8.
25. Sato S, Hirakata M, Kuwana M, et al. Autoantibodies to a 140-kd polypeptide, CADM-140, in Japanese patients with clinically amyopathic dermatomyositis. Arthritis Rheum 2005; 52:1571-6.
26. Fujisawa T, Suda T, Nakamura Y, et al. Differences in clinical features and prognosis of interstitial lung diseases between polymyositis and dermatomyositis. J Rheumatol 2005; 32:58-64.
27. Kiely PD, Chua F. Interstitial lung disease in inflammatory myopathies: clinical phenotypes and prognosis. Curr Rheumatol Rep 2013; 15:359.
28. Nakashima R, Imura Y, Kobayashi S, et al. The RIG-I-like receptor IFIH1/MDA5 is a dermatomyositis-specific autoantigen identified by the anti-CADM-140 antibody. Rheumatology (Oxford) 2010; 49:433-40.
29. Ando M, Miyazaki E, Yamasue M, et al. Successful treatment with tacrolimus of progressive interstitial pneumonia associated with amyopathic dermatomyositis refractory to cyclosporine. Clin Rheumatol 2010; 29:443-5.
30. Rutjes SA, Vree Egberts WT, Jongen P, Van Den Hoogen F, Pruijn GJ, Van Venrooij WJ. Anti-Ro52 antibodies frequently co-occur with anti-Jo-1 antibodies in sera from patients with idiopathic inflammatory myopathy. Clin Exp Immunol 1997; 109:32-40.
31. Fasano S, Gordon P, Hajji R, Loyo E, Isenberg DA. Rituximab in the treatment of inflammatory myopathies: a review. Rheumatology (Oxford) 2017; 56 :26-36.
32. Saunders P, Tsipouri V, Keir GJ, et al. Rituximab versus cyclophosphamide for the treatment of connective tissue disease-associated interstitial lung disease (RECITAL): study protocol for a randomised controlled trial. Trials 2017; 18:275.
33. Alexanderson H, Lundberg IE. The role of exercise in the rehabilitation of idiopathic inflammatory myopathies. Curr Opin Rheumatol 2005; 17:164-71.
34. Ryerson CJ, Cayou C, Topp F, et al. Pulmonary rehabilitation improves long-term outcomes in interstitial lung disease: a prospective cohort study. Respir Med 2014; 108:203-10.
35. Morisset J, Johnson C, Rich E, Collard HR, Lee JS. Management of Myositis-Related Interstitial Lung Disease. Chest 2016; 150:1118-28.
36. Kim J, Kim YW, Lee SM, Kim YS, Kim YT, Song YW. Successful lung transplantation in a patient with dermatomyositis and acute form of interstitial pneumonitis. Clin Exp Rheumatol 2009; 27:168-9.
37. Shoji T, Bando T, Fujinaga T, et al. Living-donor lobar lung transplantation for rapidly progressive interstitial pneumonia associated with clinically amyopathic dermatomyositis: report of a case. Gen Thorac Cardiovasc Surg 2013; 61:32-4.
38. Ameye H, Ruttens D, Benveniste O, Verleden GM, Wuyts WA. Is lung transplantation a valuable therapeutic option for patients with pulmonary polymyositis? Experiences from the Leuven transplant cohort. Transplant Proc 2014; 46:3147-53.
